Too Much of a Good Thing
Spoorthi Davala1, Elizabeth Gibb1, Fatima Neemuchwala1, Kelly Tuveson1, Kensho Iwanaga1,
Gywnne Church1, Ngoc Ly1
UCSF Benioff Children's Hospital Oakland, Oakland, CA, United States
Case
A 12-year-old female presented to the ED with four days of acute worsening of chronic cough and dyspnea. Upon presentation, saturations were 80% on RA and she was placed on 5LPM nasal cannula. Exam was notable for diminished breath sounds bilaterally, otherwise no crackles or wheezes. Chest x-ray showed bilateral diffuse opacification. Respiratory viral panel was positive for non-novel coronavirus 229E. She was initially admitted to the general wards, but overnight required escalation of respiratory support to 25L heated high-flow nasal canula and was transferred to ICU.
Infectious disease and Pulmonary were consulted. Further history revealed that the family lived in the Central Valley in California until she was 8 years old and she had an intermittent dry cough, improving with short acting beta agonist (SABA) as needed. Her cough resolved after moving to the Bay Area. Four months prior to presentation, she visited the Central Valley when her cough returned and did not improve with inhaled SABA and steroids.
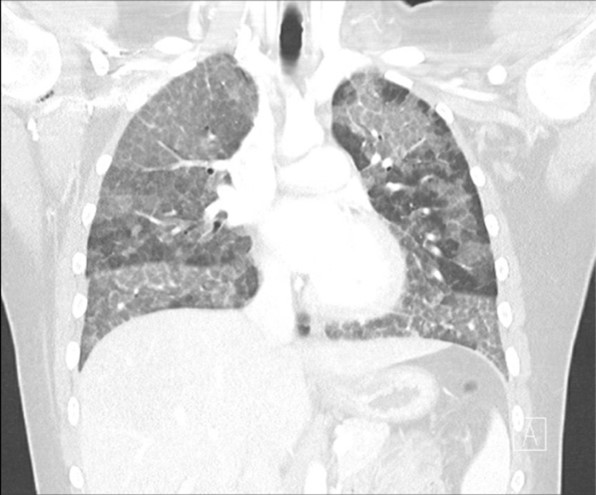
Figure 1. Chest CT was obtained.

Figure 2. Bronchoalveolar lavage was performed and showed
On the physical exam, the chest wall was symmetric, without deformity. No tenderness was appreciated upon palpation of the chest wall. The patient did not exhibit signs of respiratory distress; and normal heartbeat and breath sounds were heard upon auscultation. The patient denied chest pain, and shortness of breath.
Laboratory evaluation was unremarkable, but serology revealed that the patient was positive for Influenza type A.
Question
What is the most likely etiology of these findings?
- Coccidioides
- Mycobacterial Infection
- Bronchiolitis Obliterans
- Pulmonary Alveolar Proteinosis
- Hypersensitivity Pneumonitis
Answer D. Pulmonary Alveolar Proteinosis
Discussion
The bronchoalveolar lavage (BAL) was highly concerning for Pulmonary Alveolar Proteinosis (PAP). PAP is defined as accumulation of PAS positive lipoproteinaceous material in alveolar spaces and is caused by overproduction or inadequate clearance of surfactant. 1 The etiologies of PAP can be divided into three larger categories including autoimmune, congenital and secondary causes. Autoimmune is the most common cause and is characterized by the presence of auto-antibodies directed against GM-CSF signaling. This leads to decreased clearance of surfactant by alveolar macrophages and thus accumulation in alveolar spaces. Congenital causes present in the neonatal period and are secondary to genetic abnormalities affecting surfactant production. Secondary PAP often presents in adulthood and is caused by immunodeficiency disorders, malignancies, hematopoietic disorders and exposures to high levels of dust or toxic fumes. 1, 2
Clinically, patients can present with progressive dyspnea on exertion, cough, sputum production and fatigue. Less commonly patients may experience fever, chest pain or hemoptysis, usually in the presence of secondary infection. Physical exam findings are nonspecific, however 50% may present with crackles. Less than 25% may present with clubbing or cyanosis. 3 Routine laboratory testing are usually normal except for elevated serum lactate dehydrogenase. 1-3 Chest X-rays typically show ill-defined bilateral, symmetric air-space disease in the mid and lower lung fields. High-resolution CT scan shows ground glass opacification with intralobular and interlobular septal thickening lung tissue, often referred to as “crazy paving”. Pulmonary function testing (PFT) reveals a restrictive ventilatory defect with decreased forced vital capacity and disproportionate reduction in carbon monoxide diffusing capacity. 2
In approximately 75% of cases, BAL can be used to establish diagnosis of PAP. Lavage fluid is often described as having an opaque and milky appearance due to large, foamy alveolar macrophages filled with large amounts of periodic acid Schiff (PAS) stain-positive lipoproteinaceous material. In autoimmune PAP, serum and BAL anti-GM-CSF antibodies are elevated. To exclude other infectious etiologies, BAL fluid should be sent for cultures. If BAL findings are non-diagnostic the next step is often transbronchial or surgical lung biopsy. 1
Treatment for PAP depends on the severity and etiology of disease. In asymptomatic or mild disease, supportive care with serial monitoring of PFTs and imaging is recommended. In moderate to severe disease, whole lung lavage (WLL) continues to be the standard of care. In autoimmune PAP, several trials have shown improvement in lung function with the use of nebulized recombinant GM-CSF. 2, 3 There are also ongoing studies evaluating the role of rituximab, an anti-CD20 monoclonal antibody. 4 Other trials are evaluating the use of statins in lowering intracellular cholesterol accumulation and thus improving surfactant uptake and clearance by alveolar macrophages. 5 Therapy for congenital PAP includes supportive care and eventual lung transplant. Therapy for secondary PAP generally involves treatment of the underlying condition.
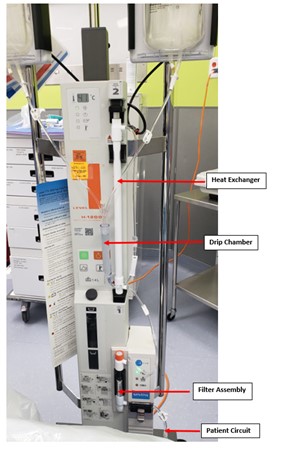
Figure 3
Multiliter bags of normal saline solution, run through a blood warmer, are used to lavage the lungs. IV tubing with a stopcock creates the lavage and drainage limbs, both of which are connected to a dual lumen endotracheal tube. Multiple drainages are performed and fluid is noted to become progressively less opaque.
Our patient’s BAL pathology report showed granular eosinophilic proteinaceous fluid, staining positive for PAS. Anti-GM CSF antibodies returned positive. Bacterial, viral and fungal cultures from BAL were negative. Autoimmune work-up was normal. She underwent a total of six WLL via rapid infuser system (see figure above) and required nocturnal positive pressure support. She was started on nebulized GM-CSF treatment (Sargramostim). Due to her inadequate response, she was then started on atorvastatin. She has subsequently improved and now remains only on statin therapy with improving PFTs. She has not required WLL since initiating atorvastatin.
References
-
Trapnell Bruce C., Whitsett Jeffrey A., Nakata Koh. (2003) Pulmonary Alveolar Proteinosis. N Engl J Med 349:26, 2527-2539.
-
Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med 2002;166:215-235
-
Goldstein LS, Kavuru MS, Curtis-McCarthy P, Christie HA, Farver C, Stoller JK. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest 1998;114:1357-1362
-
Borie R, Debray MP, Laine C, Aubier M, Crestani B. Rituximab therapy in autoimmune pulmonary alveolar proteinosis. The European respiratory journal. 2009; 33:1503–1506.
-
McCarthy C, Lee E, Bridges JP, et al. Statin as a novel pharmacotherapy of pulmonary alveolar proteinosis. Nat Commun 2018; 9:3127.


