I Know It When I See It
Hasan S. Yamin, MD (corresponding author)
Pulmonary and Critical Care Department
Nablus Specialty hospital, Nablus, Palestine
Amjad H. Abd Alhaq, MD
Pulmonary and Critical Care Department
Nablus Specialty hospital, Nablus, Palestine
Yousef Abuasbeh, MD
Thoracic Surgery Department
St. Joseph Hospital, Jerusalem, Palestine
Marwan Qabaja, MD
Clinical Pathologist
Augusta Victoria Hospital, Jerusalem, Palestine
Case
A 24 year-old male active smoker (4 Pack-years) presented with dyspnea on exertion, and dry cough of 4 months duration, he could not walk more than 100m flat ground without needing to stop to catch his breath. Physical exam showed bilateral inspiratory crackles, and significant hypoxemia SPO2 84% after climbing 2 flights of stairs. He had no previous health problems and no occupational exposures. Systemic review was negative for fever, anorexia, weight loss or arthralgias. Chest X-ray showed reticular shadows in both lungs. Echocardiography showed normal left ventricular function, normal valves, with mild pulmonary hypertension. Pulmonary function tests showed mild restriction with mild reduction in diffusion capacity. Routine chemistry was within normal limits. Chest CT was done (Figure A), followed by bronchoscopy, BAL was negative for S-100 and CD-1a.
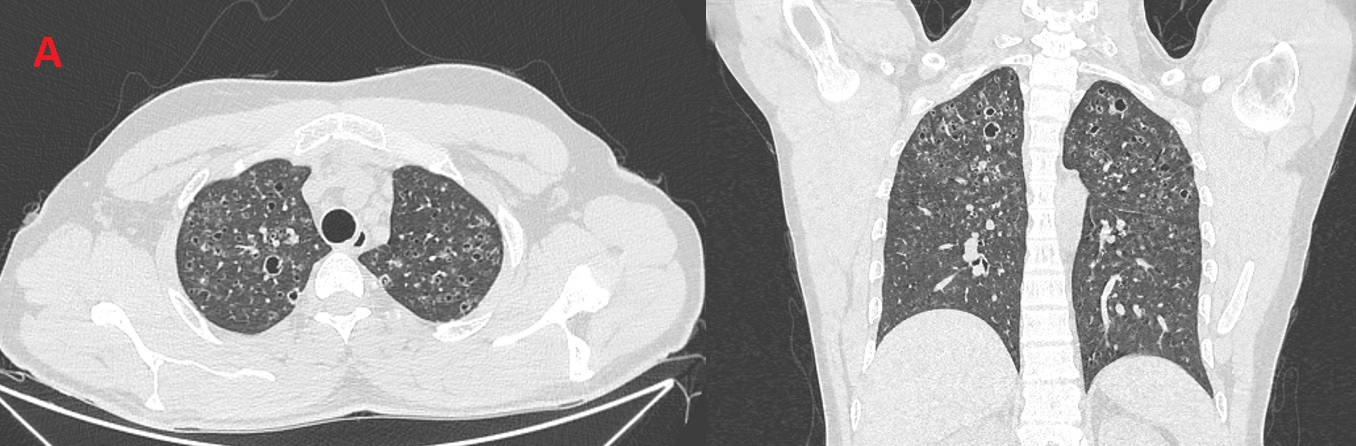
Figure (A): Bilateral upper-lobes predominant bizarre shaped cysts with scattered lung nodules.
Question
What is the most likely diagnosis?
- Metastatic malignancy
- Pulmonary Langerhans cell histiocytosis (PLCH)
- Lymphocytic interstitial pneumonitis
- Atypical infection
B. Pulmonary Langerhans cell histiocytosis
Discussion
This young male with relatively short smoking history of 4 pack- years suffers from significant dyspnea and hypoxemia on exertion, with a chest CT that is very suggestive of Pulmonary Langerhans cell histiocytosis (PLCH) showing multiple bizarre shaped cysts in mid upper zones with scattered centrilobular nodules which eventually cavitate. Pulmonary Langerhans cell histiocytosis (PLCH) is a rare cystic interstitial lung disease usually seen in young smokers in both sexes, with a peak incidence at age 20–40 years1. The pathogenesis involves cigarette smoke-induced recruitment and activation of Langerhans cells to the small airways which then activate adaptive T cell responses that lead to airway injury. Cigarette smoke may activate Langerhans cells directly, or indirectly by stimulating epithelial cells to release a variety cytokines and chemokines like granulocyte-macrophage colony-stimulating factor (GM-CSF), Chemokine (C-C motif) ligand 20 (CCL20 or Macrophage Inflammatory Protein-3 alpha), transforming growth factor-β (TGF-β), tumor necrosis factor-α (TNF-α) and osteopontin2.
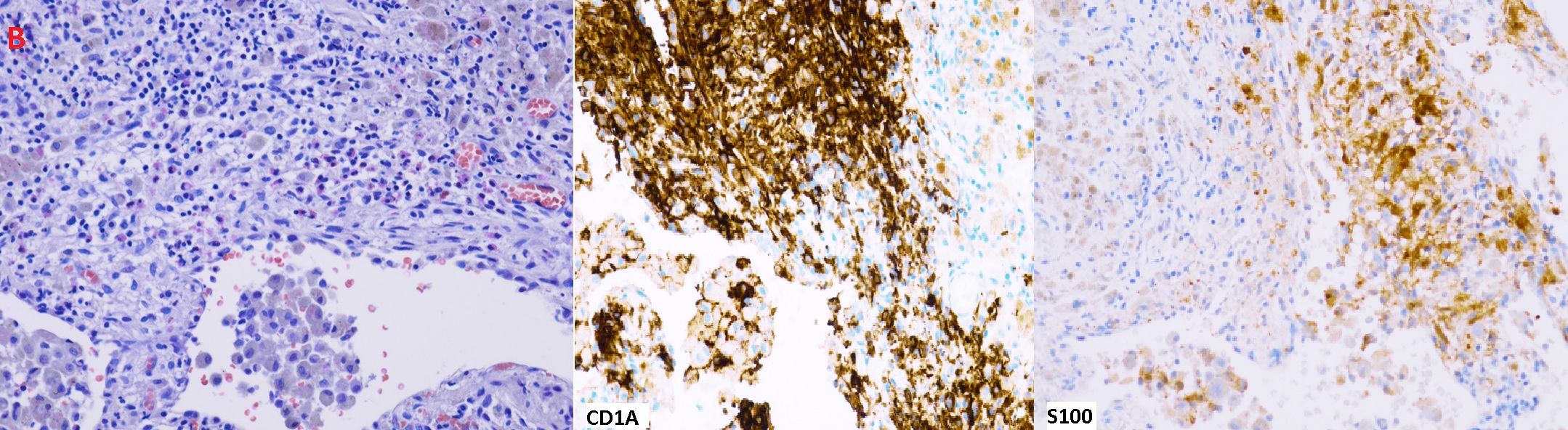
Figure (B): H&E stain: Some stellate nodules and aggregates of Langerhans cells with eosinophils. Pigmented alveolar macrophages and focal fibrosis are also seen. Langerhans cells are positive for CD1A and S100
Although the diagnosis could be made with confidence based on clinical presentation and typical CT findings, the patient opted for more diagnostic evaluation. A bronchoscopy with RUL and LUL BAL was performed. Specimen showed less than 5% CD-1a and S-100 positive cells, no malignant cells, and negative microbiologic studies. It is important to note that BAL for CD -1a is a poorly sensitive test and does not exclude the diagnosis of PLCH when negative.3,4 The diagnosis was confirmed with VATs biopsy showing infiltration of bronchioles with CD1A and S100 positive Langerhans cells (Figure B). PLCH can cause pulmonary hypertension related to a distinct vasculopathy, it is classified as group 5 in the WHO pulmonary hypertension classification.
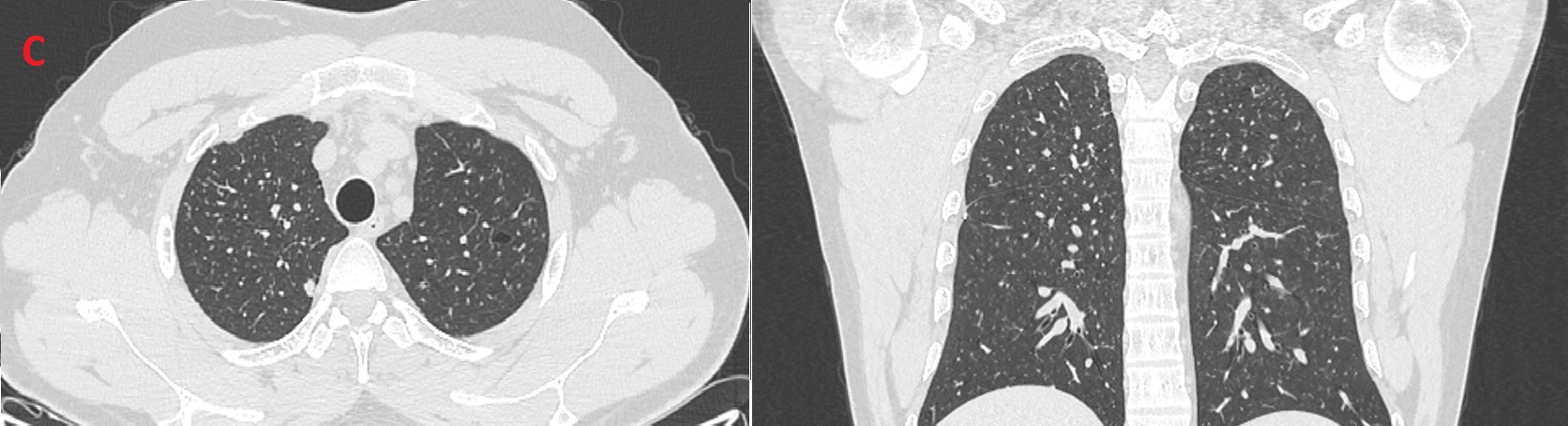
Figure (C): Follow up CT showing resolution of most previously seen bizarre shaped cysts
Patient quit smoking with aid of nicotine patches alone. Follow up after three months showed significant improvement in exercise capacity, with resolution of most previously seen bizarre shaped cysts on chest CT (Figure C), normal lung function, and reduction in pulmonary artery systolic pressure.
Other potential treatments include steroids with controversial results, Cladribine is promising in patients with progressive disease who fail to improve despite smoking cessation5. The identification of activating BRAF and mitogen-activated protein kinase (MAPK) mutations in 50% of patients opens the door for targeted tyrosine kinase inhibitors6.but these therapies are still under investigation.
Cystic pulmonary metastases should be suspected in patients with known malignancy. They tend to be predominantly basally located, multifocal, and variable in size and maybe associated with cavities. Metastatic malignancy (choice A) is unlikely in this patient with upper mid distribution of cysts and no weight loss.
Lymphocytic interstitial pneumonitis is a benign lymphoproliferative disorder limited to the lungs, more common in women than in men, and usually associated with connective tissue diseases like sjogren and rheumatoid arthritis. Cysts in Lymphocytic interstitial pneumonitis (LIP) usually have a basal predominant distribution, with associated ground glass opacities and nodules making (choice C) incorrect. Several infections might present as thin lung cysts including Pneumocystis jirovecii pneumonia, tuberculosis, non-tuberculous mycobacteria, echinococcosis among others, however this is less likely in an immunocompetent patient with no fever, and no other CT findings (ground glass opacities, lymphadenopathy) to suggest an infection (choice D).
References
-
Lorillon G, Tazi A. How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev 2017; 26: 170070
-
Suri, H.S., Yi, E.S., Nowakowski, G.S. et al. Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis 7, 16 (2012).
-
Harari S, Torre O, Cassandro R, et al. Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med 2012; 106: 1286–1292
-
Radzikowska E (2021) Update on Pulmonary Langerhans Cell Histiocytosis. Front. Med. 7:582581. doi: 10.3389/fmed.2020.582581
-
Grobost, V., Khouatra, C., Lazor, R. et al. Effectiveness of cladribine therapy in patients with pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis 9, 191 (2014). https://doi.org/10.1186/s13023-014-0191-8
-
Vassallo R, Harari S, Tazi A Current understanding and management of pulmonary Langerhans cell histiocytosis Thorax 2017;72:937-945.


