
Dyspnea and Fatigue in a Patient with Psoriasis
Rahul Khosla, MDNitin Seam, MD
Veterans Affairs Medical Center, Washington DC
History:
A 55 year old African American male presented with a four day history of fatigue, shortness of breath and lightheadedness. Over the last few hours prior to presentation, he was noted to be confused. He denied any other complaints, specifically fever, chills, cough, chest pain, orthopnea, paroxysmal nocturnal dyspnea, nausea, vomiting, melena, hematochezia or hematuria.
The past medical history was significant for severe plaque psoriasis requiring systemic therapy, non-insulin dependent diabetes mellitus, hepatitis C infection, hypertension, hypercholesterolemia, paranoid schizophrenia, obstructive sleep apnea and obesity.
Social history was notable for lack of alcohol or tobacco use and the family history was non-contributory.
Medications at time of admission were loratadine 10mg daily, aspirin 81mg daily, folic acid 1mg daily, glyburide 10 mg twice a day, triamcinolone 0.1% cream, fluocinolone acetonide 0.01% topical, diltiazem 240 mg twice a day, risperidone 6 mg nightly, calcium 500mg/vitamin D 200 U twice a day, sulfasalazine 1 gm twice a day, quetiapine fumarate 50 mg nightly, efalizumab 125mg injection once a week, metformin 1 gm twice a day, fosinopril 40 mg twice a day, prednisone 2.5mg daily and hydrochlorothiazide 25 mg daily.
He was allergic to lithium and sulfamethoxazole.
Physical Exam:
His vital signs at the time of admission showed a pulse rate of 112 / minute, respiratory rate of 22 / minute, blood pressure 120 / 71 mmHg, temperature 99.1º F and pulse oximetry on room air was 97%.
Physical examination was notable for conjunctival pallor and a II/VI non-radiating systolic murmur at the apex. The rest of the physical examination was unremarkable.
Laboratory Findings:
- WBC 13,400/cubic millimeter, differential 43 % neutrophils and 50 % lymphocytes
- Hb 3.5 g/dL, Hct 9.7% with mean corpuscular volume (MCV) 116 fL, red cell distribution width (RDW) 26%
- Plt 309,000/cubic millimeter
- PT 14 seconds, PTT 24 seconds
- Serum LDH 387 IU/L, total bilirubin 1.5 mg/dL, direct bilirubin 0.5 mg/dL and haptoglobin < 7 mg/dl.
The chest radiograph and electrocardiogram at the time of admission were unremarkable.
Question 1
What is the cause of anemia in a patient?
- Iron deficiency
- Hemolysis
- Alcoholism
- Chronic disease
- Hypothyroidism
Answer to Question 1
Answer: B
In trying to identify the cause of anemia in a patient one can adopt two general approaches:
-
- Kinetic approach1: This approach addresses the mechanism responsible for the drop in hemoglobin concentration, namely decreased RBC production, increased RBC destruction, and blood loss.
- Morphologic approach2: This approach classifies anemia according to RBC size (i.e., mean corpuscular volume) into microcytic, normocytic and macrocytic anemia.
Kinetic approach:
- Decreased RBC production: There can be numerous causes of decreased RBC production, such as:
-
- Deficiency of nutrients like folate, B12 and iron.
- Bone marrow suppression by drugs, chemotherapy, irradiation, myelodysplasia, tumor infiltration, aplastic anemia etc.
- Low levels of hormones that stimulate RBC production, such as erythropoietin (chronic renal failure), androgens (hypogonadism) and thyroid hormone (hypothyroidism).
- Anemia of chronic disease.
-
- Increased RBC destruction: Hemolysis is defined as a RBC life span of less than 100 days (normal is 120 days). Hemolysis results in anemia when the bone marrow is unable to replace more than 5 percent of the RBC mass each day.
-
- Inherited hemolytic anemias (for example, hereditary spherocytosis, sickle cell disease, thalassemia major)
- Acquired hemolytic anemias (for example, Coombs'-positive autoimmune hemolytic anemia, thrombotic thrombocytopenic purpura-hemolytic uremic syndrome)
-
- Blood loss: this is the most common cause of anemia in the industrialized world. Blood loss could be overt (for example, melena, hematochezia, trauma) or occult (for example, slow gastrointestinal hemorrhage due to peptic ulcer disease or malignancy, frequent phlebotomy).
Morphologic approach:
The normal RBC has a mean corpuscular volume of 80 to 96 femtoliters (fL) and a diameter of approximately 7 to 8 microns, equal to that of the nucleus of a small lymphocyte. Thus, RBCs larger than the nucleus of a small lymphocyte on a peripheral smear are considered large or macrocytic, while those that appear smaller are considered small or microcytic.
Macrocytic anemia:
Macrocytic anemias have a MCV above 100 fL. On a peripheral blood smear, RBCs are larger than the nucleus of a small lymphocyte. The most common causes of macrocytic anemias in the adults are (1) alcoholism, (2) liver diseases, (3) hemolysis/bleeding with reticulocytosis, (4) hypothyroidism, (5) folate or vitamin B12 deficiency, (6) exposure to chemotherapy and other drugs (hydroxyurea, zidovudine etc), and (7) myelodysplasia.
Microcytic anemia:
Microcytic anemias have small RBCs with a MCV less than 80 fl and are usually accompanied by decreased hemoglobin content within the RBC. This produces a microcytic and hypochromic appearance on the peripheral smear. The three main pathological reasons for microcytic anemias include reduced iron availability (for example, iron deficiency, chronic disease), reduced globin production (thalassemias) and reduced heme synthesis (sideroblastic anemia). The three most common causes of microcytic anemia are iron deficiency, thalassemia minor and anemia of chronic disease.
Cause of anemia in our patient:
This patient presented with an acute onset of symptoms with no obvious source of blood loss. As per the morphologic approach to diagnosing anemia this patient had macrocytosis as indicated by an elevated MCV of 116 fL. In the context of an elevated RDW, reticulocyte count and LDH level combined with low haptoglobin concentration, hemolytic anemia is the most likely etiology.
Question 2
What does the peripheral smear in figure 1 show?
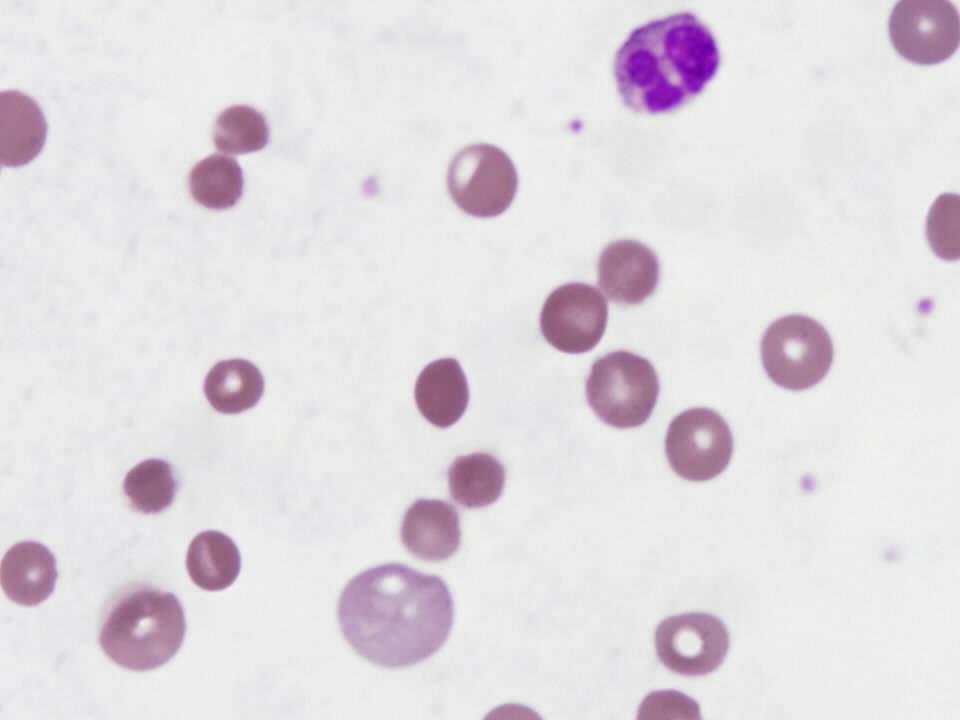
Figure 1-Patient’s peripheral smear
A. Schistocytes
B. Spherocytes
C. Spur cells
D. Sickle cells
E. Target cells
Answer to Question 2
Answer: B
Review of the peripheral smear is essential in the evaluation of suspected hemolytic anemia because it may rapidly determine the cause of RBC destruction. This smear is notable for the presence of spherocytes (see arrows in figure 2), small, dark and dense hyperchromic red blood cells without central pallor. The most likely cause of the abnormal peripheral smear in this patient is immune-mediated hemolytic anemia, though spherocytes are also seen in hereditary spherocytosis.
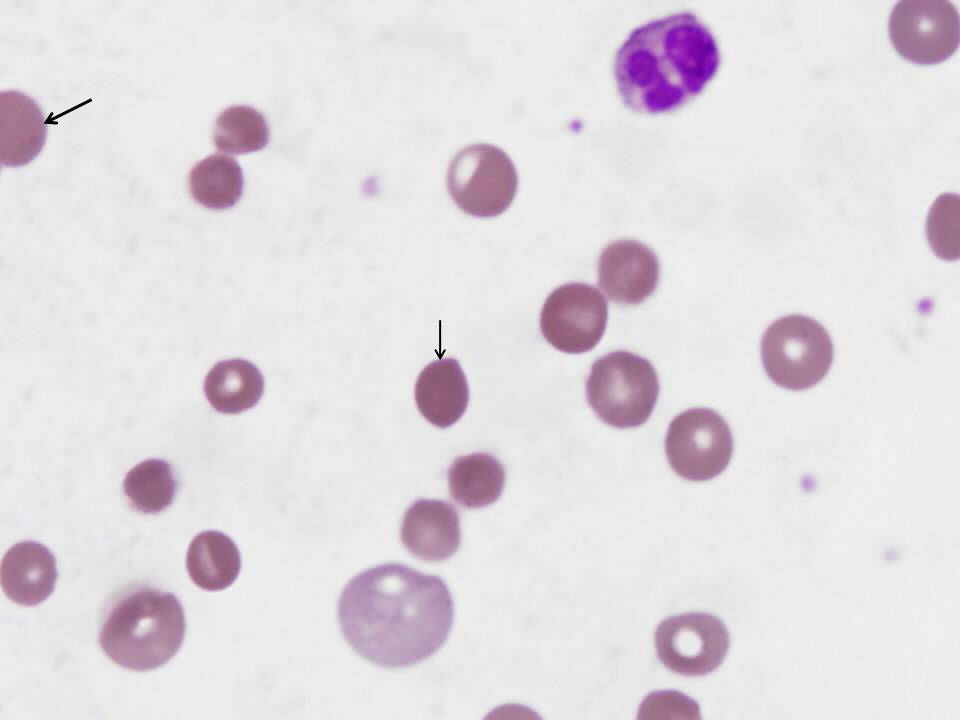
Figure 2-Peripheral smear with arrows indicating spherocytes
None of the other choices are present in the patient’s peripheral smear but if present, would suggest specific diagnoses. Schistocytes (fragmented RBCs) indicate the presence of microangiopathic hemolytic anemia and suggest life-threatening causes such as thrombotic thrombocytopenia purpura or hemolytic uremic syndrome. Normally, schistocytes comprise less than 0.5% of the RBC population. Bite cells are caused by phagocytes extracting precipitates of denatured hemoglobin. The presence of bite cells suggest oxidant damage to RBCs, which can be caused by glucose-6-phosphate deficiency or drug-induced hemolysis. Acanthocytes (spur cells) are irregular, spiculated RBCs with irregular projections, most commonly seen in patients with liver disease.3
Question 3
What is the role of antiglobulin test in evaluating hemolytic anemia?
- Confirms the presence of IgE antibodies in plasma
- Confirms the presence of autoimmune hemolytic anemia
- Confirms the absence of autoimmune hemolytic anemia
- It is useful in detecting immune-mediated hemolysis
- It is not a useful test
Answer to Question 3
Answer: D
The antiglobulin test was first described by Coombs in 1945 and is often referred to as the Coombs test.4 The test is utilized to detect the presence of erythrocyte directed IgG in the plasma or IgG/complement coating on the surface of circulating erythrocytes.
The direct antiglobulin test (DAT) is used to detect the presence of IgG /complement coating the erythrocytes. Monoclonal antihuman globulin (AHG) with specificity for IgG or various complement proteins is added to a suspension of erythrocytes. The AHG reagent is sufficiently large to cause agglutination of erythrocytes coated with IgG/complement.
DAT is useful in differentiating immune-mediated hemolysis from hemolysis caused by other factors. When DAT result is positive it is important to determine whether there is laboratory or clinical evidence of hemolysis. Laboratory evidence of hemolysis includes the presence of hemoglobinuria, elevated reticulocyte count, lactate dehydrogenase level and unconjugated bilirubin level and decreased level of hemoglobin and haptoglobin.
The indirect antiglobulin test (IAT) is used by the blood bank to detect unexpected erythrocyte antibodies in the patient’s serum or plasma. The IAT detects the presence of IgG antibodies in serum. Reagent erythrocytes are incubated in the presence of serum that potentially contains IgG antibodies. The antibodies bind to the target antigen present on the erythrocytes. After the incubation period, the erythrocytes are washed to remove unbound antibodies. Adding AHG reagent causes the IgG coated erythrocytes to agglutinate. The IAT is the final phase of the antibody screen and serologic crossmatch procedure.
There are two types of antibodies that are produced in autoimmune hemolytic anemia:
Cold agglutinins:
IgM antibodies which generally react with polysaccharide antigens on the RBC surface only at temperatures below that of the core temperature of the body. Rarely, IgG antibodies have these reaction characteristics, occurring either alone or with IgM antibodies. The DAT test is positive when anti-C3 is used but usually negative with anti-IgG.
Warm agglutinins:
IgG antibodies which generally react with protein antigens on the RBC surface at body temperature. Rarely, IgM antibodies have these reaction characteristics and readily agglutinate red cells. DAT test is invariably positive.
Question 4
Which of the following is the most likely cause of the patient’s immune-mediated hemolytic anemia?
A. Lymphoma
B. Systemic Lupus Erythematosus
C. Thrombotic thrombocytopenic purpura (TTP)
D. Efalizumab
E. Paroxysmal cold hemoglobinuria
Answer to Question 4
Answer: D
Immune hemolytic anemia describes IgG and/or IgM antibodies binding to RBC antigens and initiating RBC destruction via complement and reticuloendothelial systems. Immune hemolytic anemia has been further classified as autoimmune (production of antibodies directed against self RBCs), alloimmune (blood product transfusion exposes patient’s RBCs to allogeneic RBCs) and drug-induced.5
The patient’s positive DAT narrows the differential diagnosis to autoimmune hemolytic anemia and drug-induced hemolytic anemia. Unfortunately, these two entities are indistinguishable serologically. Warm autoimmune hemolytic anemia is often idiopathic but the most common secondary causes are lymphoproliferative and autoimmune disorders.5 Though SLE and lymphoma both cause warm autoimmune hemolytic anemia, this patient has no evidence of either of these disorders. TTP is not the correct answer because it does not cause a positive DAT test. Paroxysmal cold hemoglobinuria is not the correct answer because it is associated with cold-reactive autoantibodies, as the name implies. The correct answer is efalizumab, an anti-CD11a monoclonal antibody that is used to treat severe plaque psoriasis. Efalizumab has been reported to cause immune-mediated hemolytic anemia.6
Question 5
What is the appropriate initial treatment for this patient’s hemolytic anemia?
- Avoidance of cold
- High-dose cyclophosphamide followed by granulocyte colony-stimulating factor (G-CSF)
- Splenectomy
- Prednisone
- Cyclosporine
Answer to Question 5
Answer: D
Any possible offending drugs should be discontinued immediately and a presumptive diagnosis of drug-induced hemolytic anemia can be made if the patient responds to drug withdrawal. Corticosteroids are the initial therapy of choice for warm autoimmune hemolytic anemia and are also used in severe cases of drug-induced hemolytic anemia. Corticosteroids treat immune-mediated hemolytic anemia by decreasing antibody production. Because this patient was intermittently confused at the time of admission (a sign of severe disease) and admission hemoglobin was 3.5 g/dL, he was treated with prednisone.
A standard initial treatment regimen would be 60 to 100 mg of prednisone per day for 1 to 3 weeks.7 An initial clinical response to such a regimen should occur within 1 week and occurs in 80% of patients.8 Though case reports describe response to a variety of immunosuppressives, including cyclosporine, mycophenalate mofetil as well as cyclophosphamide followed by G-CSF, such regimens should be attempted only when corticosteroid treatment has failed or cannot be tolerated. Splenectomy is indicated in patients who fail or do not tolerate corticosteroid therapy. 50% of patients have a complete initial response to splenectomy. Avoidance of cold is not correct since it is the treatment of choice for cold agglutinin syndrome.7
References:
- Tefferi A. Anemia in adults: A contemporary approach to diagnosis. Mayo Clin Proc 2003; 78:1274.
- “Chapter 2. Clinical approach to anemia” In Hillman RS, Ault, KA (Eds) Hematology in Clinical Practice. New York, NY: McGraw Hill; 2001.
- Bull BS. “Chapter 28. Morphology of the Erythron” In Lichtman MA, Beutler E et al (Ed) Williams Hematology. New York, NY: McGraw Hill; 2006.
- Zarandona JM, Yazer MH. The role of the Coombs test in evaluating hemolysis in adults. CMAJ. 2006;174:305-7.
- Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-71.
- Kwan JM, Reese AM, Trafeli JP. Delayed autoimmune hemolytic anemia in efalizumab-treated psoriasis. J Am Acad Dermatol. 2008;58:1053-5.
- King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131-6.
- Allgood JW, Chaplin H. Idiopathic acquired autoimmune hemolytic anemia: A review of forty-seven cases treated from 1955 to 1965. Am J Med. 1967;43:254-73.


