Chronic thromboembolic pulmonary hypertension (CTEPH) is defined as a MPAP greater than 25 mmHg that persists six months following diagnosis of pulmonary embolism. The prevalence of this disorder among the pulmonary artery hypertension population is unknown. CTEPH complicates acute pulmonary embolism in two to four percent of cases. 1,2 CTEPH is thought to be vastly under-diagnosed due to the fact that many patients do not have an obvious history of pulmonary embolism. 3 It remains unclear why some patients develop CTEPH following acute pulmonary embolism and others do not. Observational studies suggest an increased risk in patients with certain hypercoagulable risk factors and chronic medical conditions.4,5,6 Pulmonary hypertension ultimately occurs as a result of macrovascular obstruction, small-vessel arteriopathy, and persistent vasoconstriction.7
Patients with CTEPH usually have a honeymoon period following the acute thromboembolism during which they are asymptomatic. Eventually, patients present with progressive dyspnea on exertion and fatigue. As pulmonary hypertension worsens and right ventricular dysfunction progresses, patients frequently develop lightheadedness, hemoptysis, chest pain, and syncope. Left untreated, death eventually occurs as a result of right ventricular failure. Relatively little is known about the natural history of CTEPH as long-term follow-up studies are lacking.
The auscultation of flow murmurs over the lung fields is a unique and specific physical exam finding not found in other types of pulmonary hypertension. These high-pitched bruits are caused by turbulent blood flow through partially obstructed pulmonary arterial branches.
This patient’s history of a mismatched ventilation/perfusion defect, possibly indicating chronic thromboembolism, is a red flag and makes idiopathic pulmonary hypertension less likely. Pulmonary veno-occlusive disease is an exceedingly rare cause of pulmonary hypertension caused by fibrotic obliteration of the pulmonary veins. These patients typically have a much more rapid progression of their disease, usually dying within two years. Additionally, unlike other causes of pulmonary hypertension, these patients typically would have evidence of pulmonary edema on imaging studies. Pulmonary hypertension can be associated with collagen vascular disease, particularly scleroderma. However, this patient’s lack of physical exam findings and symptoms consistent with collagen vascular disease make this diagnosis unlikely. Fibrosing mediastinitis is a very rare cause of pulmonary hypertension related to progressive fibrotic deposition in the mediastinum. Fibrosing mediastinitis is unlikely in this case, given that the patient’s original CT-PA failed to reveal any mediastinal calcification or compromise of other mediastinal structures including the tracheobronchial tree and esophagus.
Transthoracic echocardiography (TTE) with Doppler imaging typically provides initial evidence supporting a diagnosis of pulmonary hypertension. Echocardiographic findings include right ventricular dilatation, hypertrophy, and hypokinesis, tricuspid regurgitation (TR), as well as pressure overload as evidenced by interventricular septal deviation toward the left.8 Assessment of the TR jet allows reasonable quantification of the pulmonary arterial systolic pressure. Although sensitive for the detection of pulmonary hypertension, TTE does little to differentiate between the potential underlying etiologies in the absence of proximal pulmonary arterial thrombus, which is rarely seen.9
Ventilation-perfusion (V/Q) scanning is an effective test to differentiate chronic thromboembolism from other potential etiologies of pulmonary hypertension. A normal V/Q scan reliably excludes CTEPH. 10 In a patient with pulmonary hypertension, multiple mismatched perfusion defects on V/Q scan are highly suggestive of chronic thromboembolism as the underlying etiology. Limitations of V/Q scanning include the inability to determine the exact location, burden, and extent of disease; which is vital information in formulating the appropriate treatment plan.
CT-PA reliably identifies main and lobar pulmonary arterial clot and may provide additional information regarding anatomic localization and extent of disease. CTPA may also provide additional clues regarding the diagnosis including RV enlargement, pulmonary arterial dilation, and mosaic attenuation indicating pulmonary infarction. For these reasons CTPA is a helpful complement to V/Q scanning during the evaluation for CTEPH. Major limitations of CT-PA include poor sensitivity for segmental and distal disease as well as poor ability to differentiate acute thrombus from chronic, endothelialized thrombus. CTPA has been compared to V/Q scanning in the identification of chronic thromboembolism and has been shown to be significantly less sensitive.10 For this reason, CT-PA is not the most appropriate initial test to exclude CTEPH.
Pulmonary angioscopy involves the use of a fiberoptic scope for intraluminal pulmonary arterial visualization to the segmental level. Pulmonary angioscopy has largely been replaced by advancements in non-invasive imaging techniques. It still has potential value in operative planning for patients diagnosed with CTEPH; however, it would not be the initial test of choice.
Our patient initially had an echocardiogram revealing moderate right ventricular enlargement, moderate right atrial enlargement, and moderate TR; with an estimated pulmonary artery systolic pressure of 75 mmHg. V/Q lung scan showed a large mismatched defect in the apical segment of the right lung, a moderate mismatched defect in the apical segment of the left lung, and multiple small perfusion defects in the left lower lung base and superior segment of the left upper lobe (Figure 1,2).
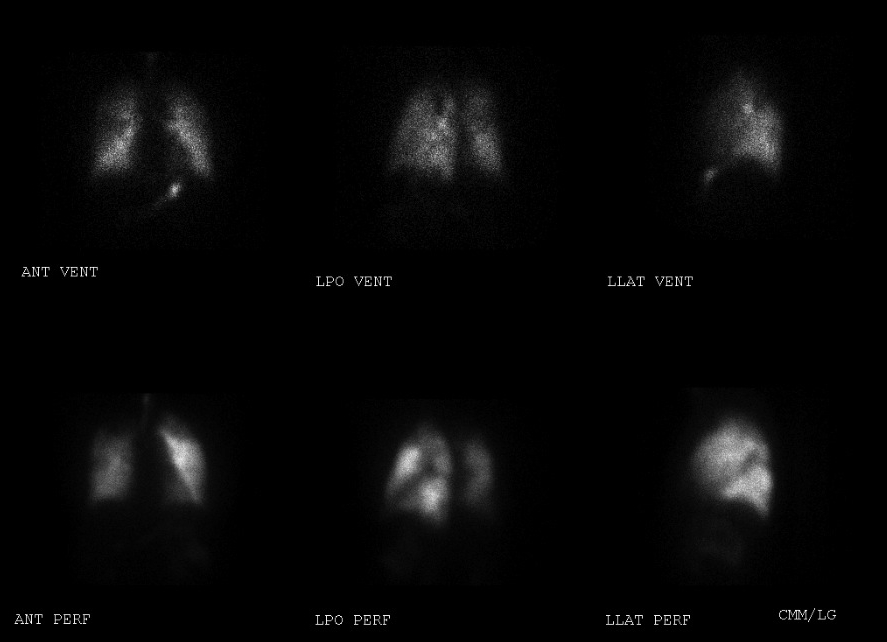
Figure 1. Ventilation/Perfusion scan images of the left lung revealing mismatched defects in the apical segment of the left lung, superior segment of the left lower lobe, and the left lower lung base.
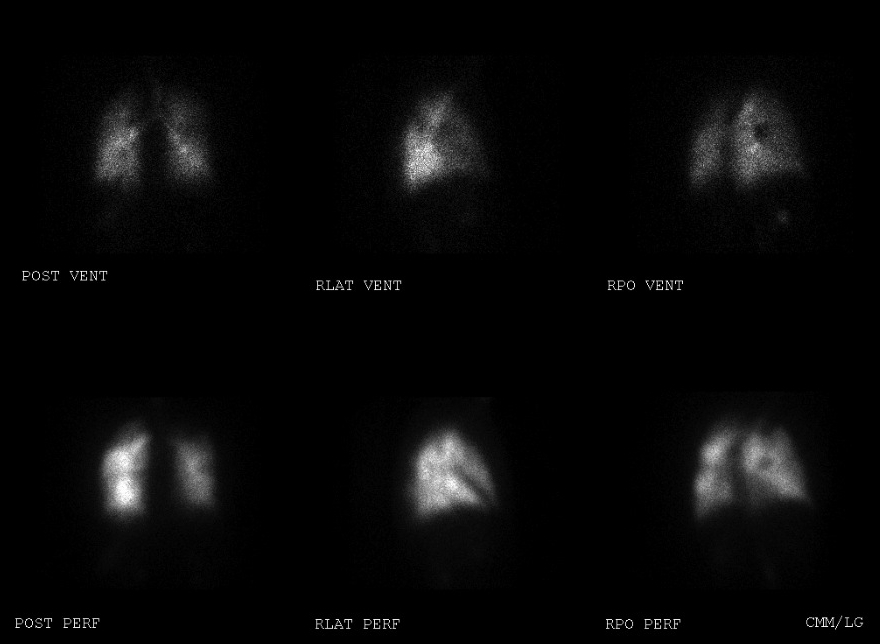
Figure 2. Ventilation/Perfusion scan images of the right lung revealing a large mismatched defect in the apical segment of the right lung.
Pulmonary angiography and right heart catheterization are the gold standard in the diagnosis of CTEPH and the assessment of operability. Angiography reliably differentiates chronic thromboembolic disease from the thrombus of acute pulmonary embolism. Angiographic patterns associated with chronic thromboembolic disease include vascular webs or bands, intimal irregularities, abrupt narrowing of the main pulmonary arteries, and obstruction of lobar or segmental vessels at their origin.11
Right heart catheterization provides a precise, quantitative hemodynamic assessment of the degree of pulmonary hypertension. In addition, concomitant vasodilator responsiveness can be assessed which provides both prognostic information and can guide medical therapy in patients ultimately deemed inoperable.
Patients with risk factors for coronary artery disease undergo coronary angiography prior to thromboendarterectomy. Coronary artery bypass grafting (CABG) can then be performed if necessary at the time of the thromboendarterectomy during the re-warming phase.12
Percutaneous transfemoral inferior vena cava (IVC) filter insertion is usually performed prior to surgery to prevent recurrent thromboembolism.13 Data to support this practice is lacking.
Our patient had pulmonary angiogram performed revealing non-acute pulmonary embolus in the anterior segment of the left upper lobe, the anterior basal segment of the left lower lobe, the apical segment of the right upper lobe, and the medial basal and posterior basal segments of the right lower lobe. Simultaneous right heart catheterization yielded severe pulmonary arterial hypertension with a MPAP of 45 mmHg and a normal PCWP of 7 mmHg. Coronary angiography revealed an isolated 30% stenosis of the left main coronary artery, further evaluated with intravascular ultrasound and found to have no compromise of flow.
The treatment of choice for CTEPH is pulmonary thromboendarterectomy. This procedure involves median sternotomy with cardiopulmonary bypass and periods of circulatory arrest during which the endothelialized clot is carefully dissected away from the underlying intima. Following successful thromboendarterectomy, patients immediately develop drastic reductions in pulmonary arterial pressure and pulmonary vascular resistance and improvement in cardiac output.14 Hemodynamic improvements result in reverse right ventricular remodeling and return of normal right ventricular systolic and diastolic function. These physiologic changes result in significant improvements in functional capacity as evidenced by 6-minute walk distances and New York Heart Association (NYHA) class.15 Benefits have been shown to persist for at least 9-12 months. Long-term mortality due to CTEPH following pulmonary endarterectomy is low among patients who survive three- months post-operatively; approximately one percent at one year and six percent at three years.
Medical management is considered only in patients with inoperable disease, those with recurrent pulmonary hypertension following thromboendarterectomy, and as bridging therapy to surgery. Epoprotenol infusion, prostacyclin analogues, endothelin receptor antagonists, and phosphodiesterase-5 inhibitors have all been used with modest degrees of success in CTEPH patients deemed non-operative and those with residual pulmonary hypertension following thromboendarterectomy.16
Our patient underwent IVC filter placement and pulmonary thromboendarterectomy at an experienced center. Bosentan was continued in the preoperative period as bridging therapy. During the procedure, a large amount of obstructive, hard, white clot was carefully dissected away from the right and left pulmonary vasculature (Figure 3).

Figure 3. Chronic thromboemboli removed from this patient by thromboendarterectomy. Extension of the thromboemboli into the lobar and segmental branches is visible.
Following removal, intra-operative trans-esophageal echocardiography (TEE) revealed immediate resolution of paradoxical ventricular septal motion and tricuspid regurgitation.
Thirty-day mortality following pulmonary endarterectomy has been estimated to be between 5-10% and appears to be dependent on the level of expertise of the performing center. Most postoperative mortality is related to reperfusion pulmonary edema and residual pulmonary hypertension. Pulmonary artery steal syndrome is a phenomenon caused by redistribution of blood flow to newly patent pulmonary vasculature away from the previously well-perfused segments.17
Reperfusion pulmonary edema is an entity related to acute lung injury in those segments of lung which are newly reperfused.18 When combined with pulmonary artery steal, life-threatening and prolonged respiratory failure can result as blood is redirected towards acutely damaged, noncompliant, poorly ventilated segments of lung.
Patients are maintained on lifelong anticoagulation with warfarin following surgery. Repeat thromboembolism can occur in these patients despite systemic anticoagulation and IVC filter placement; although this occurs in less than 0.5% of patients. Repeat pulmonary thromboendarterectomy has been successfully employed in this patient population. 19
Residual pulmonary hypertension is an important predictor of late mortality. Although significant improvements in hemodynamics and functional capacity may be seen; pulmonary thromboendarterectomy is not a curative procedure in patients with distal, surgically-inaccessible thromboemboli and in those with severe, diffuse small-vessel arteriopathy related to long-standing severe pulmonary hypertension. These patients may be candidates for advanced medical therapy with vasodilator agents as discussed above. Patients who progress to end-stage, severe pulmonary hypertension may be candidates for lung transplantation.
Our patient had an uncomplicated post-operative course and her Bosentan was discontinued. She is asymptomatic, NYHA class I, six months following surgery. Follow-up TTE revealed normal RV size and function with a significantly improved PA systolic pressure of 25 mmHg. She will be maintained on warfarin indefinitely.


